Example: Dynamics in the Bose-Hubbard Model¶
We want to simulate the Bose-Hubbard model, and look at some basic dynamics. So first we write our Hamiltonian that defines this model.
(5)¶![\hat{H} = -t \sum_{\langle i, j \rangle}
\left[\hat{b}_i^{\dagger} \hat{b}_j + \mathrm{h.c.} \right]
+ U \sum_{i} \frac{\hat{n}_i \left(\hat{n}_i - 1 \right)}{2}
- \mu \sum \limits_i \hat{n}_i](../_images/math/8e9205feb8aa4dc608369db0cef2f8250c39baed.png)
Equation (5) is the general expression for the Hamiltonian of the Bose-Hubbard model. For this exercise we work in the canonical ensemble (fixed particle number), and we ignore the chemical potential and write the Bose-Hubbard model as Eq. (6).
(6)¶![\hat{H} = -t \sum_{\langle i,j\rangle}
\left[\hat{b}_i^{\dagger} \hat{b}_j + \mathrm{h.c.} \right]
+ U \sum_{i} \frac{\hat{n}_i \left(\hat{n}_i - 1 \right)}{2}](../_images/math/d5959e9d7b16b989eb6211970781c5192ec0e3c3.png)
Here we have our creation and annihilation operators,
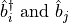 , and our number operator,
, and our number operator,
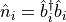 , as usual. Additionally,
our second term is scaled by the parameter that describes site interactions,
, as usual. Additionally,
our second term is scaled by the parameter that describes site interactions,
 .
.
First we must include the necessary python libraries. The two that are
needed are the MPSPyLib, which includes how python processes the MPS matrix
inputs and format the user’s Hamiltonian into the proper format for the
Fortran compiler to run. The post-process flag is an argument in the main
function and can be passed as command line argument as --PostProcess=F
or --PostProcess=F. We have imported
two different libraries, and use the handles mps and np to refer to functions
within each library. Thus any time a function is preceded with one of these
handles, (i.e. Ops.BuildBoseOperators())
1import MPSPyLib as mps
2import numpy as np
3from sys import version_info
4import matplotlib.pyplot as plt
5import sys
Next we need to create which operators we want to observe in our Hamiltonian.
So for the Bose-Hubbard model, we want bosonic operators, so we call
Ops.BuildBoseOperators(), with a maximum of 5 particles allowed on
a given lattice site. This is the term inside the parentheses. We then want
build a custom operator: we are choosing to look at our interaction term,
which will be defined as
 . The
following lines correspond to creating this operator, and setting up our
system.
. The
following lines correspond to creating this operator, and setting up our
system.
24 # Build operators
25 Operators = mps.BuildBoseOperators(5)
26 Operators['interaction'] = 0.5 * (np.dot(Operators['nbtotal'],
27 Operators['nbtotal'])
28 - Operators['nbtotal'])
Then we build our Hamiltonian. We need to add two terms (or two sums) to our
Hamiltonian. So we first define a variable that will act as our Hamiltonian,
which in this case will be H.
30 # Define Hamiltonian MPO
31 H = mps.MPO(Operators)
32 H.AddMPOTerm('bond', ['bdagger', 'b'], hparam='t', weight=-1.0)
33 H.AddMPOTerm('site', 'interaction', hparam='U', weight=1.0)
Note that we have two different types of MPO terms present in our Hamiltonian
(here we have 'bond' and 'site' terms). This is because the summations
for each term in the Hamiltonian are different. The bond summation considers
nearest-neighbor interactions, and has different site indices,
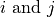 , for each operator. MPS takes each operator fed to
the function (in this case the
, for each operator. MPS takes each operator fed to
the function (in this case the 'bdagger', 'b' expression) and assigns
indices in the order which it appeared in the list. The other type of summation
is simply over all of the sites. This type of MPO term expects only one operator
(or a single expression of operators).
Next we define what observables we want to measure in our ground state. We can look at the number operator at each site
35 # Ground state observables
36 myObservables = mps.Observables(Operators)
37 # Site terms
38 myObservables.AddObservable('site', 'nbtotal', 'n')
39 # Correlation functions
40 myObservables.AddObservable('corr', ['nbtotal','nbtotal'], 'nn')
41 myObservables.AddObservable('corr', ['bdagger','b'], 'spdm')
Then we want to define our correlation operators when looks at neighboring
sites. We define two observables, 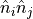 and
and
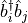 . This is done by the following
commands:
. This is done by the following
commands:
40 myObservables.AddObservable('corr', ['nbtotal','nbtotal'], 'nn')
41 myObservables.AddObservable('corr', ['bdagger','b'], 'spdm')
Next we set the convergence parameters for our simulation. It is important to set the convergence for finding our ground state to a reasonable tolerance to get an accurate ground state. Note: this can be optimized along with simulation run times.
43 # Convergence parameters
44 myConv = mps.MPSConvParam(max_bond_dimension=20, max_num_sweeps=6)
45 myConv.AddModifiedConvergenceParameters(0, ['max_bond_dimension',
46 'local_tol'], [50, 1E-14])
47
48 myDynConv = mps.TDVPConvParam(max_num_lanczos_iter=20,
49 lanczos_tol=1E-6)
We have modified our convergence parameters to check that our max bond
dimension in our system is no larger than 50 and our local tolerance is
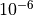 . We set our Krylov-Convergence parameters to do a maximum
of 20 Lanczos Iterations with a tolerance of for each
calculation. It should be noted that computation time is largely impacted
by these convergence parameters. If tolerances are set too small for a given
system, computation time can be greatly increased.
. We set our Krylov-Convergence parameters to do a maximum
of 20 Lanczos Iterations with a tolerance of for each
calculation. It should be noted that computation time is largely impacted
by these convergence parameters. If tolerances are set too small for a given
system, computation time can be greatly increased.
In this simulation we also define a second set of observables to show that we can look at dynamics for different portions of our simulation. Here we want to look at the number operator for each site as our system is evolving. So we define a new observable dictionary:
51 # Dynamics time evolution observables
52 dynObservables = mps.Observables(Operators)
53 # Site terms
54 dynObservables.AddObservable('site', 'nbtotal', name='n')
The next step is to define values for our Hamiltonian we created, assign what
location and name our simulation output will be run with, and define what
type of simulation we want to run. We first define our parameters
and 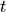 in our Hamiltonian. We also define the number of lattice sites.
in our Hamiltonian. We also define the number of lattice sites.
56 # Specify constants and parameter lists
57 U = 10.0
58 t = 1.0
59 staticsParameters = []
60 L = 6
61 tlist = [5.0, 20.0]
Then we want to run a simulation over two different variations, where the
value of U is changed at different rates. This will define our quenching
function which will allow us to ramp our function value . This is
done through:
63 for tau in tlist:
64 # Quench function ramping down
65 def Ufuncdown(t, tau=tau):
66 return 10.0 + 2.0 * (1.0 - 10.0) * t / tau
67 # Quench function ramping back up
68 def Ufuncup(t, tau=tau):
69 return 1.0 + 2.0 * (10.0 - 1.0) * (t - 0.5 * tau) / tau
70
71 Quenches = mps.QuenchList(H)
72 Quenches.AddQuench(['U'], 0.5 * tau, min(0.5 * tau / 100.0, 0.1),
73 [Ufuncdown], ConvergenceParameters=myDynConv)
74 Quenches.AddQuench(['U'], 0.5 * tau, min(0.5 * tau / 100.0, 0.1),
75 [Ufuncup], ConvergenceParameters=myDynConv)
76
77 staticsParameters.append({
78 'simtype' : 'Finite',
79 # Directories
80 'job_ID' : 'Bose_Hubbard_mod',
81 'unique_ID' : 'tau_'+str(tau),
82 'Write_Directory' : 'TMP_03/',
83 'Output_Directory' : 'OUTPUTS_03/',
84 # System size and Hamiltonian parameters
85 'L' : L,
86 't' : t,
87 'U' : U,
88 # Specification of symmetries and good quantum numbers
89 'Abelian_generators' : ['nbtotal'],
90 'Abelian_quantum_numbers' : [L],
91 # Convergence parameters
92 'verbose' : 1,
93 'logfile' : True,
94 'MPSObservables' : myObservables,
95 'MPSConvergenceParameters' : myConv,
96 'Quenches' : Quenches,
97 'DynamicsObservables' : dynObservables
98 })
In this looping structure, we define our quenches, which will change the value
of one of our parameters, in this case .
71 Quenches = mps.QuenchList(H)
72 Quenches.AddQuench(['U'], 0.5 * tau, min(0.5 * tau / 100.0, 0.1),
73 [Ufuncdown], ConvergenceParameters=myDynConv)
74 Quenches.AddQuench(['U'], 0.5 * tau, min(0.5 * tau / 100.0, 0.1),
75 [Ufuncup], ConvergenceParameters=myDynConv)
We note that Dynamics.AddQuench() takes the Hamiltonian, the
variable in our Hamiltonian which we want to ramp, the range of values for
which we will apply this function, assigns a function which calculates the
new value, and the set of convergence parameters that we want to abide by.
Then we append these parameters in the form of a dictionary list to be used for our simulations.
77 staticsParameters.append({
78 'simtype' : 'Finite',
79 # Directories
80 'job_ID' : 'Bose_Hubbard_mod',
81 'unique_ID' : 'tau_'+str(tau),
82 'Write_Directory' : 'TMP_03/',
83 'Output_Directory' : 'OUTPUTS_03/',
84 # System size and Hamiltonian parameters
85 'L' : L,
86 't' : t,
87 'U' : U,
88 # Specification of symmetries and good quantum numbers
89 'Abelian_generators' : ['nbtotal'],
90 'Abelian_quantum_numbers' : [L],
91 # Convergence parameters
92 'verbose' : 1,
93 'logfile' : True,
94 'MPSObservables' : myObservables,
95 'MPSConvergenceParameters' : myConv,
96 'Quenches' : Quenches,
97 'DynamicsObservables' : dynObservables
98 })
Here we have three different sections of assignments. We have our simulation
references, 'job\_ID' which is what each simulation file will be named,
with a unique ending given by the value assigned to the key 'unique\_ID'.
Then we assign where these files should be saved. We want our compilation
files to be saved in the local folder TMP_03 and all of our outputs to the
local folder OUTPUTS_03.
Next we define our system. We need to call every unassigned variable in our
Hamiltonian, and also specify our system size. We do this through the key
assignments for 'L', our system size, 't' and 'U' the variables
discussed in our Hamiltonian in Eq. (6).
Next we have the section which we can define any symmetries or quantum
numbers for our system. These are set by the keys 'Abelian_generators'
and 'Abelian_quantum_numbers'.
And finally we describe which convergence parameters we want to use for each section of our analysis, and which observables we want to measure for each portion of our analysis. This is done through the lines 94-97.
Next we have the primary function calls that convert this python dictionary
to the fortran files that will be used to actually run the MPS algorithms.
This is done by calling the dictionary we just defined, and passing the
operators, the Hamiltonian, and the option to post process data for our
simulation. Then we call the function tools.runMPS() to run the
simulation.
101 MainFiles = mps.WriteFiles(staticsParameters, Operators, H,
102 PostProcess=PostProcess)
Then we run two simulations (from the parameter list we defined above), and read both the statics data and the dynamics data back into the local python process.
105 if(not PostProcess):
106 if os.path.isfile('./Execute_MPSMain'):
107 RunDir = './'
108 else:
109 RunDir = None
110 mps.runMPS(MainFiles, RunDir=RunDir)
111 return
112
113 # Postprocessing and plotting
114 # ---------------------------
115
116 Outputs = mps.ReadStaticObservables(staticsParameters)
117 DynOutputs = mps.ReadDynamicObservables(staticsParameters)
In the last part we collected already the information of the Losschmidt echo over time. The remaining lines plot the overlap with the initial state over time for the two different quench times:
135 plt.rc('font', family='serif')
136 plt.rc('mathtext', fontset='cm')
137 plt.plot(np.linspace(0, 1, len(le_list[0])), le_list[0], 'r-',
138 label="tq=5")
139 plt.plot(np.linspace(0, 1, len(le_list[1])), le_list[1], 'g-',
140 label="tq=20")
141 plt.xlabel(r"Time in percent " r"$t / t_q$", fontsize=16)
142 plt.ylabel(r"Loschmidt Echo "
143 r"$| \langle \psi(t) | \psi(0) \rangle |^2$",
144 fontsize=16)
145 plt.legend(loc="lower left")
146 if(ShowPlots):
147 plt.savefig('03_BoseHubbardDynamics.pdf', bbox_inches='tight')
148 plt.show()
149
150 return